 />i
/>iA private equity client asked us recently to assess a rumor that FDA was on the warpath in enforcing the 510(k) requirement on medical devices from a particular region. Such a government initiative would significantly deter investments in the companies doing the importing. Turns out, the agency was not. The FDA’s recent activities in the region were well within their historical norms.
But the project got us thinking, what does the agency’s enormous database on import actions tell us about the agency’s enforcement priorities more generally? There are literally thousands of ways to slice and dice the import data set for insights, but we picked just one as an example. We wanted to assess, globally, over the last 20 years, in which therapeutic areas has FDA been enforcing the 510(k) requirement most often?
Background
FDA maintains and publicly releases a database of import inspection activity that includes import refusals. The database contains much information for each refusal, including the establishment registration number of the importing firm, name, address, product description, a shipment ID, the clinical area in which the product is classified, industry of importer and a code representing the reason for FDA’s refusal. FDA supplies in a downloadable excel spreadsheet a complete list of those codes and what they represent.
Over the last 22 years reflected in the database, there have been approximately 125,000 refusals of device shipments.
Methodology
There is a specific code, 508, that identifies those refusals made based on a lack of a required 510(k). To be sure, FDA can have multiple reasons for refusing entry. Thus, I filtered the data on the basis that at least one of those reasons was 508. There were a little over 18,000 such refusals over the last 22 years. We could just as easily do one for the absence of premarket approvals, but we did not combine them because we were afraid it would obscure trends related specifically to the 510(k) requirement.
Visualized Findings
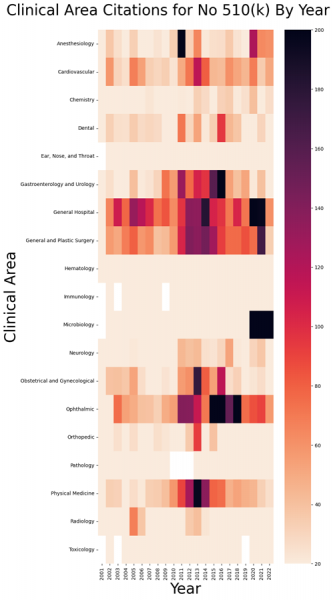
We decided to present the results in a heat map showing where, in the context of import refusals, FDA activity enforcing the 510(k) requirement has been greatest over time and in relation to clinical area.
Interpretation
I find the visualization fascinating in that there are quite a few import refusals based on the lack of a 510(k), and wide differences by clinical area and over time for those refusals.
Predicted Volume of FDA Refusals
Mechanically, Customers and Border Protection (CBP) uses an automated system to screen the data entered for entry. If the import contains a medical device, data will be sent to FDA and their PREDICT system[1], as seen in Figure 1. The system will query databases for relevant information such as facility registrations, drug listings, market approvals, etc. Source data may include field exams, sample analyses, results of foreign and domestic facility inspections, product code and facility code accuracy, data anomalies in transmitted information, and admissibility history (importer, exporter, manufacturer and ultimate consignee). [2] If accurate data are provided, the entry will pass the new system auto look-up and likely be recorded as “May Proceed” by the system. If the entry does not pass auto look-up, it will be manually reviewed by an Entry Reviewer, as shown in Figure 2. As a result, it’s important for importers to fill out the paperwork properly so as not to get snared by the automated system. It’s possible that much of what we are seeing is simply the product of incorrect paperwork that ultimately gets straightened out manually.
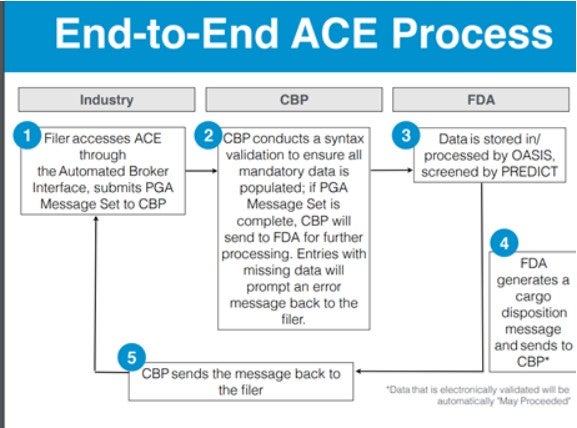
Figure 1: Automated Commercial Environment (ACE) Process
Most importantly, if the entry gets kicked out of the automated system, to assess whether a 510(k) is needed, inspectors look at the product code that the importer declared for the product and look up in FDA’s database to see whether a 510(k) is required, and then look to the paperwork supplied by the importer to see whether a 510(k) for such a product code is identified. That 510(k) simply needs to be associated with the importer’s facility establishment number. Thus, when the automated system refuses entry, FDA can look for when the import paperwork does not identify any 510(k) when, based on the claimed product code, a 510(k) is required.
In some ways, import enforcement in this space is difficult for FDA because it is partly built on an honor system. The import paperwork needs to identify an applicable 510(k) and device listing for the products being imported, if a 510(k) is required. But the import inspector really has little opportunity to assess whether that assertion is valid or not.
It is not the most elegant or refined analysis, but it does catch those importers who are referencing a product code that requires a 510(k) but do not have it listed with their establishment. The fact is no one should be caught at the border by the system if they simply looked up in advance whether the product code they are using requires a 510(k). It’s simple to do on FDA’s website.
The FDA import inspectors have very little visibility into whether the claimed 510(k) is applicable to the product or not. For example, if a product has been modified, such that a new 510(k) would be required but was incorrectly documented as an internal letter to file rather than a new 510(k), the FDA staff at the ports have little ability to assess that. All they can really check is whether in the system the importer has a 510(k) for the product code which the company has declared for the product being imported.
Unfortunately, dishonest or unknowing importers might simply claim that their product fits within a product code that does not require a 510(k), and the inspectors might not notice. A lot of freight moves through ports, and these inspectors do not have the time or the technical background to go through and physically inspect all of the products and their instructions for use, and make a judgment about what the appropriate product code is. Further, the inspectors really have no infrastructure or system in place, as already explained, to make a judgment about whether the referenced 510(k) is up-to-date with regard to the current design of the product.
You might be tempted to assume that people know whether a 510(k) is required for their imported product, and dishonest people take measures to avoid getting caught. If the system is mostly just catching a few honest people who did not realize a 510(k) is required for a given product code, and is not catching people who are shrewd (dishonest) enough to at least fill out the paperwork in a way that passes the inspection, you would expect enforcement to be overall light in volume.
While the data can’t tell us how many crooks are out there getting away with importing an unapproved product, apparently there are a lot of honest people out there who apparently just haven’t checked whether their product code requires a 510(k). To FDA’s credit, the system is keeping those products from being imported. It’s just surprising to me that this many people haven’t done the basic checking, once they found a product code, to figure out whether a 510(k) is required.
IVDs
We did notice one high level trend. It appears that those areas in the chart that are distinctly low in terms of FDA enforcement levels on this particular issue of no 510(k) are in vitro diagnostic devices, or IVD’s. In the chart, this includes chemistry, hematology, immunology, microbiology, pathology and toxicology. Compare the enforcement levels there with general hospital or general surgery.
We asked ourselves why? To be sure, with IVD’s, many of them are exempt from 510(k), but many are not. Many IVD’s are Class I devices that nonetheless do require a 510(k). Unfortunately, many people think that a Class I product means you do not have to submit a 510(k) at all. Further, some of international products might be considered components of IVD’s rather than finished IVD’s, or they might be placed in categories such as research use only. But honestly, we did not come up with a satisfactory theory to explain why IVD’s are not refused entry more often on the basis of no 510(k) compared to other clinical areas.
Other Comparisons
Aside from that high-level trend, it’s also interesting to simply compare two different clinical areas, such as ophthalmology with orthopedics. Both include many products that require a 510(k), and yet ophthalmology devices tend to have much more difficulty at the border with the 510(k) issue.
FOOTNOTES
[2] http://www.aaei.org/wp-content/uploads/2015/12/FDA-Best-Practices-Manual-Update-FINAL-Sept-2015.pdf
Dawn Norman also contributed to this article.