

 />i
/>iThe FDA recently released a final guidance, titled “510(k) Third Party Review Program” and known as 3P510k, discussing the factors the FDA uses to determine whether a Class I or Class II medical device is eligible for review by a third party and also how third parties can become accredited to perform 510(k) reviews.
In addition, this final guidance outlines the FDA's process for the recognition, suspension, and withdrawal of recognition for 3P510k review organizations, and explains how those organizations can ensure consistent quality of work.
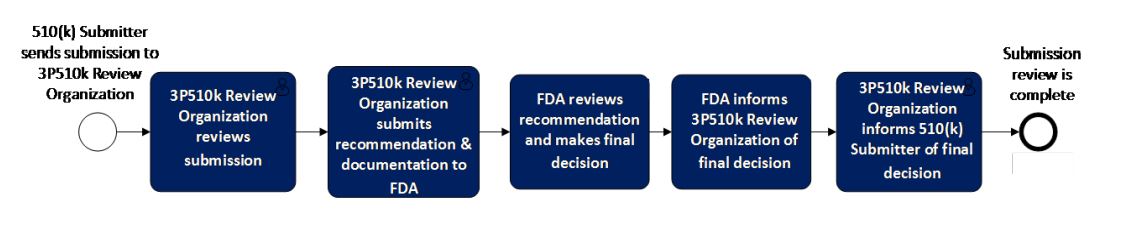
The guidance includes the following diagram summarizing the 3P510k program:
This guidance also outlines the FDA’s current thinking on key aspects of the 3P510k Review Program, including:
- Expectations for 3P510k organization reviews of 510(k) submissions, including the policy for early interaction consults
- Requirements and recommendations for recognition and re-recognition of 3P510k review organizations under the 3P510k program
- Content and format of a 3P510k organization’s application for initial recognition and re-recognition
- Process for suspension or withdrawal of recognition
- Leveraging the International Medical Device Regulators Forum’s requirements for Regulatory Reviewers under the Good Regulatory Review Practices and the Medical Device Single Audit Program, as appropriate
In Section V, the guidance discusses six factors the FDA will consider in determining device type eligibility for the 3P510k Review Program.
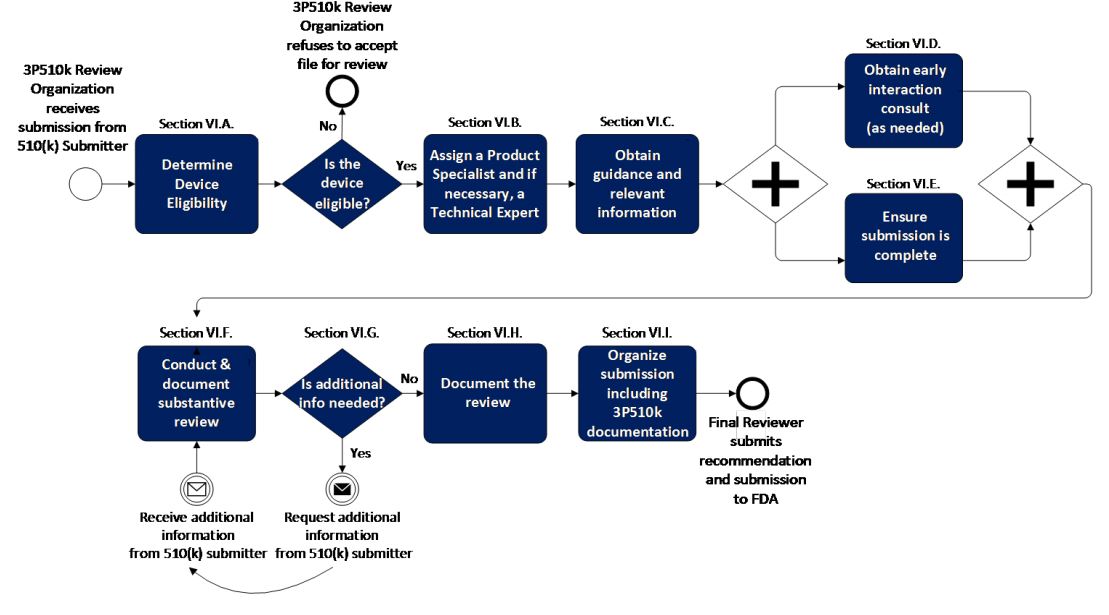
Section VI describes the manner in which 3P510k review organizations should conduct their review of 510(k)s, as summarized in the graphic below:
The FDA describes the criteria used to decide which 3P510k review organizations are qualified to conduct premarket reviews of eligible 510(k)s in Section VII of the guidance.
In Section VIII of the guidance, the FDA provides recommendations on what third parties should include in an application to the FDA for recognition as a 3P510k review organization.